



Clinical description of patients with adult-onset Still’s disease: case series from a rheumatology center in Guatemala
https://doi.org/10.46856/grp.10.e173
Cite as:
Bran Ordóñez A, Guevara Mejía JE, Bautista Esquivel YD, García Kutzbach A. Descripción clínica de pacientes con enfermedad de Still del adulto, serie de casos en un centro reumatológico guatemalteco. Global Rheumatology. Vol 4 / En - Jun [2023] Available from: https://doi.org/10.46856/grp.10.e173
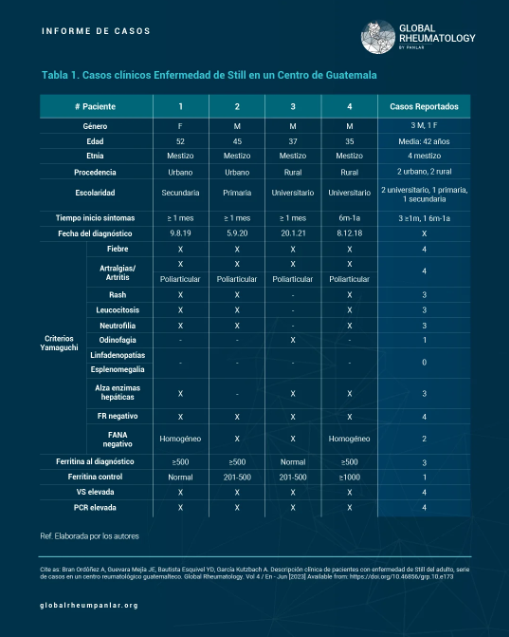
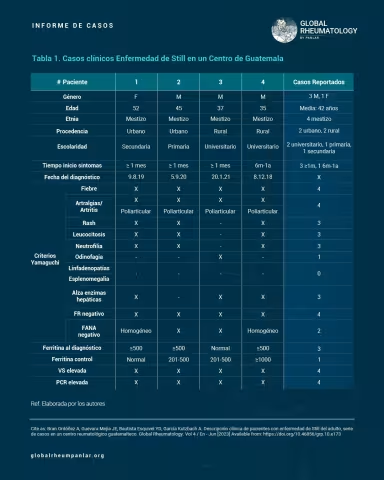
Tabla 1 - Descripción clínica de pacientes con enfermedad de Still del adulto, reporte de 4 casos en un centro reumatológico Guatemalteco
{kind=link}
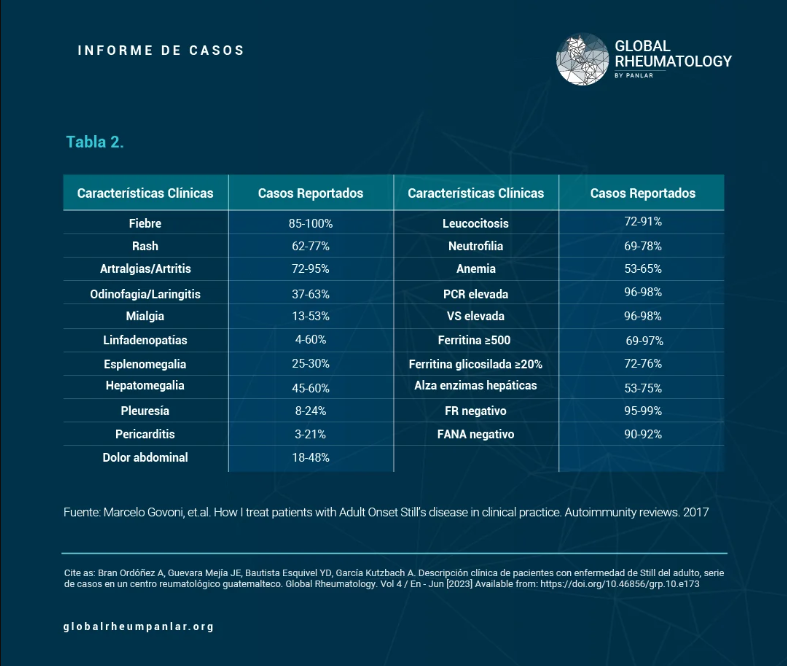
Tabla 2 - Descripción clínica de pacientes con enfermedad de Still del adulto, reporte de 4 casos en un centro reumatológico
{kind=link}
Carta comité de ética
1184 Views
License
This is an open-access article distributed by the terms of the Creative Common Attribution License (CC-BY NC-4). The use, distribution or reproduction in other forms is permitted, provided the original author(a) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with this terms.
Clinical description of patients with adult-onset Still’s disease: case series from a rheumatology center in Guatemala
Introduction: Still's disease is a rare autoinflammatory disorder that affects mostly young adults, with an unknown cause and difficult to diagnose. This study describes four cases of adult-onset Still's disease in Guatemala, providing valuable information for future research.
Methodology: A descriptive study was conducted based on patient records from 2021 to 2022. A questionnaire was designed to gather patient information.
Results: Four patients (3 males, 1 female) with a mean age of 42 were included in the study. The most common symptoms were fever and joint pain, with all patients exhibiting leukocytosis, neutrophilia, and elevated liver enzymes. Two patients had positive antinuclear antibodies. Three patients had ferritin levels between 500-1000 ng/ml, while one patient had levels over 1000 ng/dl. All patients received steroids and methotrexate, with one patient also receiving leflunomide due to inadequate response. Adherence to treatment varied, with two patients attending regular appointments and two not.
Conclusions: This is the first description of adult-onset Still's disease in Central America, providing valuable information for future research. Ferritin levels are useful markers of inflammation and may aid in the optimization of steroid treatment. The majority of patients adhered well to treatment and follow-up, highlighting the importance of consistent medical care for this rare disease.
Key Messages
- This is the first case description of adult-onset Still's disease in the Central American region, specifically in Guatemala to our knowledge.
- Still's disease is a rare and underdiagnosed autoimmune pathology.
- Most patients had good adherence to treatment and follow-up appointments, which is crucial for disease control.
- Further research is needed to improve early diagnosis and better treatment, as well as to monitor promising new treatments with biologics.
Adult-onset Still’s disease was first described by George F. Still in 1897, based on 22 cases of children with chronic polyarthritis (1). In 1971, Eric Bywaters described 14 similar cases with adult-onset presentation (1,11,14,23,25). It is a rare multisystemic autoinflammatory disorder of unknown etiology (2,4), slightly more frequent in women than men, and more common among young adults between 19 and 35 years of age, though cases have also been reported in the elderly (>70 years) (5,6). Its incidence in the United States is estimated at 1–3 cases per million (3), while in Colombia it is estimated at 0.16–0.4 cases per 10,000 inhabitants (27). It is considered one of the most challenging febrile illnesses to diagnose due to the absence of a specific diagnostic test (3). The disease has been associated with HLA B17, B18, and DR2 (29), a multigenic pattern (5,6,29), and various infectious agents, mostly viral (26), which may trigger the disease in genetically predisposed individuals (26,29).
The clinical hallmark of the disease is daily spiking fever for 1–2 days exceeding 39°C, typically in the evening, which resolves spontaneously and is accompanied by a transient salmon-pink macular/maculopapular rash. The rash commonly appears on the trunk, extremities, palms, soles, and face. Other manifestations include sore throat, oligoarticular and transient arthralgia/arthritis, which may progress to a more severe form—symmetrical polyarthritis—affecting wrists, knees, ankles, elbows, shoulders, hand joints, and the temporomandibular joint. Generalized myalgia, cervical lymphadenopathy, hepatosplenomegaly, and serositis are also frequent features (4,5,6,23,27,28). Interleukins 1, 1β, 2, 6, 8, 18 and tumor necrosis factor alpha (TNF-⍺) play a key role in these manifestations (5,6,29).
Less frequent signs of severe or advanced disease include myocarditis, cardiac tamponade, diffuse alveolar hemorrhage, pulmonary hypertension, aseptic meningitis, uveitis, amyloidosis, disseminated intravascular coagulation, and macrophage activation syndrome (5,6).
Common laboratory abnormalities include neutrophilic leukocytosis, elevated liver enzymes, and high levels of acute-phase reactants (erythrocyte sedimentation rate [ESR], C-reactive protein [CRP], and ferritin). Less frequently, anemia of chronic disease, thrombocytosis, negative antinuclear antibodies (ANA) and rheumatoid factor (RF), and complement consumption may be found (5,6). The disease can lead to complications such as transient pulmonary hypertension, macrophage activation syndrome, diffuse alveolar hemorrhage, thrombotic thrombocytopenic purpura, and amyloidosis, all of which reduce life expectancy (5,6,23,27).
Two classification criteria are used: Yamaguchi and Fautrel. However, Yamaguchi's criteria are more widely used due to the ease of obtaining the necessary laboratory tests, whereas Fautrel’s require glycosylated ferritin, which is less accessible (5,6). Yamaguchi’s criteria are divided into major and minor: major criteria include fever ≥ 39°C, arthralgia/arthritis > 2 weeks, transient macular/maculopapular rash, leukocytosis ≥ 10,000/μl with > 80% neutrophils; minor criteria include sore throat, significant lymphadenopathy, hepatosplenomegaly, abnormal liver function (AST/ALT and LDH), and negative ANA and RF. The disease is classified if the patient meets ≥ 5 criteria, including at least 2 major criteria (5,6). Diagnosis is primarily clinical and by exclusion (7), and treatment involves anti-inflammatories, corticosteroids, disease-modifying antirheumatic drugs (DMARDs), and biological agents (3,5,6).
Four patients diagnosed with Adult-onset Still’s Disease were included, all of whom were seen in outpatient consultations and presented for evaluation over the course of one year (February 2021 to February 2022). Data were collected through direct clinical interviews and analysis of their medical records, using a questionnaire specifically designed for this purpose. Initially, the 1997 Yamaguchi classification criteria (7) were assessed, and during follow-up visits, treatments were documented.
Data were collected in Excel, where they were reviewed and analyzed using 2x2 tables and graphs. Descriptive statistics were applied. Data confidentiality was ensured, and the ethical principles established by the Declaration of Helsinki were respected, with informed consent obtained from all participants.
Image
There is no Guatemalan literature on this disease; however, there are multiple international articles regarding its presentation, pathogenesis, clinical markers, evolution, and treatment (27). In Latin America, there is more information in Brazil, Argentina, Chile, and Peru; only in recent years has it started to be described and followed in Colombian literature, with the publication of retrospective studies and case series mainly analyzing treatment response, pathophysiology, and diagnosis (27).
In this case series, four patients were analyzed: three men and one woman, with a male-to-female ratio of 4:1. Regarding ethnicity, all four were mestizos, and the educational level was predominantly university (two patients). Age ranged from 35 to 52 years, consistent with the article by Mathieu Gerfaud-Valentin and collaborators, who reported that it usually affects young adults (9), and Álvaro José Muriel R. et al., who described ages between 28 and 36 years (young adults) (28).
Using the Yamaguchi classification criteria, the most frequently observed features were: fever, arthralgia/arthritis, and negative rheumatoid factor (RF), present in all four patients. Evanescent rash, leukocytosis, neutrophilia, and elevated liver enzymes were found in three patients (75%) (Table 2), as reported in the study by Govoni et al. (Table 2) (12). Antinuclear antibodies (ANA) were negative in two patients; however, in that same study, 90–92% of patients showed ANA negativity (Table 2) (12). Additionally, no patients presented with lymphadenopathy and/or splenomegaly, differing from the findings of Govoni et al., who reported that approximately 30% of their patients had these symptoms (Table 2) (12). Fever and arthralgia were the most common symptoms, also consistent with the study by Santos Castañeda et al. (10).
In Colombia, Álvaro José Muriel R. et al. (28) described the main manifestations as fever (95%), characteristic skin rash, and joint involvement (86%); similarly, half had odynophagia, and 30% had palpable lymph nodes. In this series, one patient presented with hepatomegaly and transient liver function test abnormalities (28).
In the study by Anis and Chan (16), they reported unusual forms of presentation: recurrent pleuritis and pericarditis (20–30%), and absence of typical symptoms except for fever (16). Similar findings were noted in the study by Govoni et al., in which 8–24% of patients had pleuritis and 3–21% had pericarditis (12). None of the patients in this clinic presented with these findings. In the study by Bodard et al., 61% had pericarditis (16), twice the rate reported by Govoni et al. This finding has become increasingly frequent, making echocardiography important.
In the study by Ruscitti et al. (22), it was noted that pulmonary disease may suggest an emerging cause of mortality in this condition, as already observed in juvenile forms (22).
Of the four patients studied, three sought care for the first time after more than one year of symptom progression, and only one had symptoms for 6 months to one year.
Ferritin plays a role in various conditions, including chronic diseases, neurodegenerative disorders, and malignancies (11).
Ferritin levels were assessed at the time of diagnosis and during follow-up, with three patients showing values between 500–1000 ng/mL at diagnosis, and one patient with 1000 ng/mL. These levels are similar to those found in the study by Govoni et al., who reported >500 ng/mL in 69–97% of cases (12). Serum ferritin is considered an essential diagnostic tool for this disease. A threshold of five times the normal value (1000 µg/L) is considered suggestive, with 40.8% sensitivity and 80% specificity; it correlates not only with disease activity but also with severity (21).
Follow-up ferritin levels showed that two patients were between 201–500 ng/mL after six months of treatment, one patient still had very high levels (>1000 ng/mL), and one had normalized.
All clinic patients received steroids, consistent with the study by Piero Ruscitti and collaborators, with an average dose of 20 mg/day, which could be withdrawn in 50% of cases within 3 to 6 months (8). Another Colombian study published by Catalina Quilindo et al. (26) reported the use of prednisone at a dose of 0.5 mg/kg with significant response; these drugs are used together with non-steroidal anti-inflammatory drugs (NSAIDs) as first-line treatment (26). This shows that cases in Latin America and Europe are managed similarly.
All patients received methotrexate concomitantly. There was no need for biological agents in the clinic; however, the following biologics have been studied and may be useful, especially in refractory cases: anti-IL-1 agents such as anakinra (canakinumab or rilonacept), which are effective steroid-sparing and disease-modifying agents, leading to remission and proving superior to conventional therapy (14).
Comorbidities and adverse effects included immune thrombocytopenic purpura, osteoporosis, and hypertension.
In follow-up, three patients continued on methotrexate, one on leflunomide (due to inadequate response to methotrexate), and one on steroids due to active disease.
Regarding treatment adherence, two patients regularly attended follow-up appointments, and two had not returned for about six months. Three patients remained asymptomatic, without disease relapse, and one patient continued to experience polyarticular pain after 1 year and 5 months of treatment.
None of the studied patients developed disease-related complications; however, some have been reported in other studies, such as marantic endocarditis, described in the Peruvian study by Horacio Suárez-Ale and Sandra Solís-Torres (30). This reinforces the importance of educating patients to seek medical attention at the first sign of symptoms in order to avoid such complications and protect their lives.
This is the first case series description of adult-onset Still's disease in the Central American region, representing a relevant contribution to future investigations and clinical references in the regional context.
Adult-onset Still’s disease is a rare autoinflammatory condition and is often underdiagnosed. In this series, most patients met all of Yamaguchi’s major criteria, which facilitated timely diagnosis and allowed for early initiation of treatment.
Ferritin, recognized as an inflammatory marker, remained persistently elevated in some cases despite treatment, highlighting its usefulness not only as a diagnostic tool but also as a guide for optimizing corticosteroid therapy.
It is worth noting that most patients showed good adherence to treatment and clinical follow-up, which is essential for keeping the disease under control and preventing long-term complications.
1. Nalini Valluru, et. Al. Rare manifestation of a rare disease, acute liver failure in adult onset Still’s disease: dramatic response to methylprednisolone pulse therapy – a case report and review. Hindawi Publishing Corporation, Case reports in medicine. 4 de junio de 2014; 2014 (375035)
2. Katerina Damevska MD, PhD. Et.al. Adult-onset Still’s disease as a cutaneous marker of systemic disease. Clinics in dermatology. 2019; 2019 (37): 668-674.
3. Sabeeda Kadvath, Petros Efthimiu. Adult-onset Still’s disease-pathogenesis, clinical manifestation, and new treatment options. ANN MED. 2015; 2015 (47): 6-14.
4. William Medina-Chamaidán, Dr. Francisdco Medina-Morataya MD. Fiebre, poliartritis y exanema, enfermedad de Still del Adulto. The Ecuador Journal of medicine. 2020; 2020 (185): 58-71.
5. Elena Riera Alonso, Alejandro Olivé Marqués. 2019. Adult-onset Still disease (173, 1437-1443). Marc. C. Hochberg. et. al. Rheumatology, 7th edition. Elsevier.
6. Duane pearson. Adult -onset still’s disease. Sterling West, et.al, Rheumatology secrets, 2019, chapter 24, 209-212
7. Philippe Guilpain, Alain Le Quellec. About the complexity of adult onset Still-s disease… and advances still required for its management. BMC Medicine. 2017; 2017 (15): 5.
8. Duane Pearson MD. Adult-onset Still Disease. En: Radjan Lourde Selvanadin/Marybeth Thiel/Angie Breckon. Rheumatology secrets. Fourth edition. Philadelpia: Elsevier; 2020. 209-213.
9. Mathieu Gerfaud-Valentin, et. Al. Adult-onset Still’s disease. Autoimmunity Reviews. 2014; 2014 (13): 708-722.
10. Santos Castañeda, et. Al. Adult-onset Still’s disease: Advances in the treatment. Clinical rheumatology. 2016; 1-17.
11. Cristina Rosário, et. Al. The hyperferrtinemic Syndrome: macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC medicine, 2013; 2013 (11); 185.
12. Marcelo Govoni, et.al. How I treat patients with Adult Onset Still’s disease in clinical practice. Autoimmunity reviews. 2017; 2017 (16): 1016-1023.
13. Tomoyuki Asano, et. Al. Adding colchicine to immunosuppressive treatments; a potential option for biologic-refractory adult-onset Still’s disease. BMC research Notes. 2018; (11): 320.
14. Guido Junge, June Mason, Eugene Feist. Adult onset Still’s disease – The evidence that anti-interleukin-1 treatment is effective and well-tolerated (a comprehensive literature review). Seminars in arthritis and rheumatism. 2017; (47): 295-302.
15. Arsany Anis, MD and Kok Hoe Chan, MD. An unusual presentation of Adult-onset Still’s disease in a patient with recurrent pleural and pericardial effusions. Am J Med Sci. 2021; 361 (5): 655-658.
16. Quentin Bodard, et. Al. Cardiac involvement in adult-onset still’s disease: Manifestations, treatments and outcomes in a retrospective study of 28 patients. Journal of autoimmunity. 2021; (116): 102541.
17. Piero Ruscitti, Tanja A Stamm, Roberto Giabomelli. Changing the outcome measures, changing the results? The urgent need of a specific disease activity score to adult-onset Still’s disease. Anna Rheum Dis, BMJ/eular. 21 de mayo de 2020; (0):0.
18. Yuya Fujita, et. Al. Elevated serum levels of checkpoint molecules in patients with adult Still’s disease. Arthritis research & Therapy. 2020; (22):174.
19. Carlo Umberto Mazini, et. Al. Elevated troponin serum levels in Adult Onset Still’s disease. Hindawi publishing corporation, case report in rheumatology. 30 de enero de 2015; 732095.
20. Stéphane Mitrovic and Bruno Fautrel. New markers for Adult-onset Still’s disease. Joint bone spine. 18 de mayo de 2017; (2018) 285-293.
21. Piero Ruscitti, et. Al. Parenchymal lung disease in adult onset Still’s disease: an emergent marker of disease severity-characterization and predictive factor from Gruppo italiano di Ricerca in Thraumatologia Clinica e Sperimentale (GIRRCS) cohort of patients. Arthritis research & Therapy, BMC, 2020; (22): 151.
22. Serena Colafrancesco, Roberta Priori and Guido Valesini. Presentation and diagnosis of adult-onset Still’s disease: the implications of current and emerging markers in overcoming the diagnostic challenge. Expert Rev. Clin. Immunol. 2015; 11(6), 749-761.
23. Bella Y. Mehta, et. Al. Racial/ethnic variations in morbidity and mortality in Adult Onset Still’s disease: an analysis of national dataset. Seminars in arthritis and rheumatism. 2019; 469-473.
24. Umut Kalyoncu, et. Al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still’s disease: Data from a large multicenter cohort. Journal of autoimmunity. 2016; XXX (2016):1-5.
25. D. Lebrun, et. Al. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semnars in Arthritis & Rheumatism. 2017; (17): 30129-4.
26. Quilindo C, Morales K, Guerrero A. Enfermedad de Still, un diagnóstico diferencial importante: Reporte de un Caso. RFS Rev Fac Salud [Internet]. 2017 [citado el 15 de noviembre de 2022];9(1):21–5. Disponible en: https://journalusco.edu.co/index.php/rfs/article/view/1931
27. Prieto-Torres AE, Suárez-Molina W, Pantoja-Agreda JI. Adult Onset Still's Disease (AOSD): A rare condition with a classic presentation. Case Report. Case rep [Internet]. 2020;6(2):100–8. Disponible en: http://dx.doi.org/10.15446/cr.v6n2.83482
28. Muriel R. ÁJ, Rueda G. JM, González Buriticá H, Castaño C. O. Una patología poco frecuente: la enfermedad de Still del adulto. Experiencia clínica con 17 casos. Rev Colomb Reumatol [Internet]. 2016;23(2):126–30. Disponible en: http://dx.doi.org/10.1016/j.rcreu.2016.01.003
29. Peruilh L, Tapia G, Petit-Breuilh V, Valenzuela F, Carreño L. Enfermedad de Still del adulto, a propósito de un caso: Un desafío diagnóstico. Rev chil dermatol [Internet]. 2018;32(4). Disponible en: http://dx.doi.org/10.31879/rcderm.v32i4.127
30. Suárez-Ale H, Solís-Torres S. Enfermedad de Still del adulto asociada a endocarditis marántica: reporte clínico y revisión de literatura. Rev Soc Peru Med Interna (línea) [Internet]. 2015 [citado el 15 de noviembre de 2022];28(1):18–24. Disponible en: https://revistamedicinainterna.net/index.php/spmi/article/view/176